Dispersion interactions between neighboring Bi atoms in (BiH3)2 and Te(BiR2)2
Rebekka Haack; Stephan Schulz; Georg Jansen
文献索引:10.1002/jcc.25209
全文:HTML全文
摘要
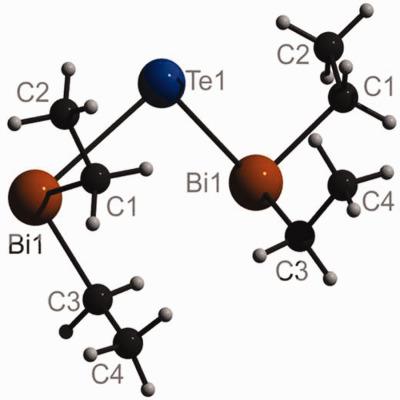
Triggered by the observation of a short Bi⋯Bi distance and a BiTeBi bond angle of only 86.6° in the crystal structure of bis(diethylbismuthanyl)tellurane quantum chemical computations on interactions between neighboring Bi atoms in Te(BiR2)2 molecules (R = H, Me, Et) and in (BiH3)2 were undertaken. Bi⋯Bi distances atoms were found to significantly shorten upon inclusion of the d shells of the heavy metal atoms into the electron correlation treatment, and it was confirmed that interaction energies from spin component‐scaled second‐order Møller–Plesset theory (SCS‐MP2) agree well with coupled‐cluster singles and doubles theory including perturbative triples (CCSD(T)). Density functional theory‐based symmetry‐adapted perturbation theory (DFT‐SAPT) was used to study the anisotropy of the interplay of dispersion attraction and steric repulsion between the Bi atoms. Finally, geometries and relative stabilities of syn–syn and syn–anti conformers of Te(BiR2)2 (R = H, Me, Et) and interconversion barriers between them were computed. © 2018 Wiley Periodicals, Inc.