| Structure | Name/CAS No. | Articles |
|---|---|---|
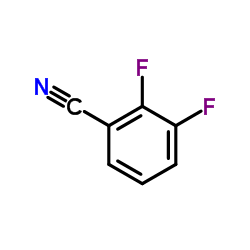 |
2,3-Difluorobenzonitrile
CAS:21524-39-0 |
| Structure | Name/CAS No. | Articles |
|---|---|---|
|
|
2,3-Difluorobenzonitrile
CAS:21524-39-0 |