Accurate lattice energies of organic molecular crystals from periodic turbomole calculations
Hannes Konrad Buchholz; Matthias Stein
文献索引:10.1002/jcc.25205
全文:HTML全文
摘要
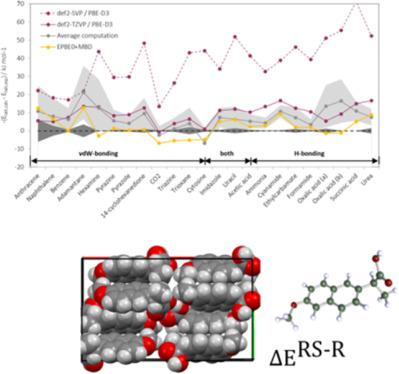
Accurate lattice energies of organic crystals are important i.e. for the pharmaceutical industry. Periodic DFT calculations with atom‐centered Gaussian basis functions with the Turbomole program are used to calculate lattice energies for several non‐covalently bound organic molecular crystals. The accuracy and convergence of results with basis set size and k‐space sampling from periodic calculations is evaluated for the two reference molecules benzoic acid and naphthalene. For the X23 benchmark set of small molecular crystals accurate lattice energies are obtained using the PBE‐D3 functional. In particular for hydrogen‐bonded systems, a sufficiently large basis set is required. The calculated lattice energy differences between enantiopure and racemic crystal forms for a prototype set of chiral molecules are in good agreement with experimental results and allow the rationalization and computer‐aided design of chiral separation processes. © 2018 Wiley Periodicals, Inc.