
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:PF-477736
- 纯度:98.0%
- 规格:5mg/25mg/1g/1g
- 联系人:阿拉丁

 2881459338
复制
2881459338
复制
- 联系电话:400-620-6333
- 长沙福臻生物科技有限公司
- 湖南省 长沙市 芙蓉区
- 产品名:(alphaR)-alpha-氨基-N-[5,6-二氢-2-(1-甲基-1H-吡唑-4-基)-6-氧代-1H-吡咯并[4,3,2-ef][2,3]苯并二氮杂卓-8-基]环己烷乙酰胺
- 纯度:98.0%
- 规格:500mg/1g/5g/10g
- 联系人:张经理
3756953817
复制

 18229892877
复制
18229892877
复制

- 联系电话:18229892877
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:PF-477736
- 纯度:98.0%
- 规格:
- 联系人:徐经理
351666998
复制
 133 1186 9306
复制
133 1186 9306
复制

- 联系电话:133 1186 9306
查看所有供应商和价格请点击:
952021-60-2
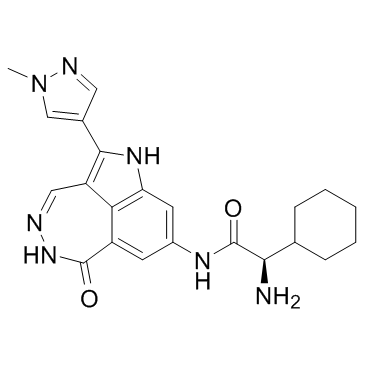
952021-60-2结构式
| 中文名 | (alphaR)-alpha-氨基-N-[5,6-二氢-2-(1-甲基-1H-吡唑-4-基)-6-氧代-1H-吡咯并[4,3,2-ef][2,3]苯并二氮杂卓-8-基]环己烷乙酰胺 |
|---|---|
| 英文名 | Cyclohexaneacetamide, α-amino-N-[5,6-dihydro-2-(1-methyl-1H-pyrazol-4-yl)-6-oxo-1H-pyrrolo[4,3,2-ef][2,3]benzodiazepin-8-yl]-, (αR) |
| 英文别名 |
(2R)-2-Amino-2-cyclohexyl-N-[2-(1-methyl-1H-pyrazol-4-yl)-6-oxo-5,6-dihydro-1H-[1,2]diazepino[4,5,6-cd]indol-8-yl]acetamide
PF-477736 PF 477736 |
| 描述 | PF 477736 是一种有效的,ATP 竞争性的 Chk1 抑制剂,Ki 为 0.49 nM,对其选择性是 Chk2 (Ki, 47 nM) 的 100 倍。 |
|---|---|
| 相关类别 | |
| 靶点 |
Chk1:0.49 nM (Ki) Chk2:47 nM (Ki) CDK1:9.9 μM (Ki) |
| 体外研究 | PF 477736(PF-00477736,0.01-1μM)在CA46细胞中以剂量依赖性方式消除喜树碱诱导的DNA损伤检查点。如通过细胞存活测定所确定的,PF 477736(180,360,540nM)在HT29细胞中以时间和剂量依赖性方式增强吉西他滨诱导的细胞毒性。 PF 477736(360 nM)与吉西他滨联合使用可显着降低CDK1的磷酸化,这与检查点消除和细胞进入有丝分裂一致[1]。 PF 477736(PF-00477736)对CDK1活性的抑制作用很差,Ki为9.9μM[1] [2]。 |
| 体内研究 | PF 477736(PF-00477736,4-60mg/kg,ip)每天一次(sid)或每天两次(bid)治疗单独在人结肠Colo205异种移植模型中表现出无抗肿瘤活性。然而,PF 477736依赖性地增强了吉西他滨MTD的抗肿瘤活性。 PF 477736(15和30 mg/kg)诱导组蛋白H3磷酸化和DNA损伤,并增加来自体内异种移植模型的Colo205肿瘤样品中的细胞凋亡[1]。 |
| 细胞实验 | 用吉西他滨(15nM)或喜树碱(25nM)处理HT29或人脐静脉内皮细胞16小时。随后以不同浓度添加PF 477736(PF-00477736)。在加入PF477736后4至48小时,除去含药物的培养基并将细胞在无药物培养基中培养。当载体处理的对照细胞90%汇合时(接种8天),收获细胞并通过Coulter计数器计数[1]。 |
| 动物实验 | 当肿瘤在指定的治疗方案中体积为100至150mm 3时,通过ip注射施用化学治疗剂或PF 477736(PF-00477736)。吉西他滨在一系列剂量下给药,包括小鼠的最大耐受剂量(MTD),根据每3天一次进行四次治疗(q3d×4)时间表。 PF477736在吉西他滨后24小时开始根据q3d×4方案施用一系列剂量(4-60mg / kg)。考虑到腹膜内给药和体重减轻的行为反应的严重程度为5%至10%,PF 477736的MTD确定为40mg / kg。对于细胞毒性药物,MTD的平均体重减轻的发生率为5%~10%[1]。 |
| 参考文献 |
| 密度 | 1.6±0.1 g/cm3 |
|---|---|
| 分子式 | C22H25N7O2 |
| 分子量 | 419.480 |
| 精确质量 | 419.206970 |
| PSA | 134.48000 |
| LogP | 0.95 |
| 外观性状 | yellow |
| 折射率 | 1.790 |
| 储存条件 | 2-8°C |
| 水溶解性 | DMSO: ≥20mg/mL |
| 危险品运输编码 | NONH for all modes of transport |
|---|