L-368,899 hydrochloride
Modify Date: 2025-08-25 09:53:02

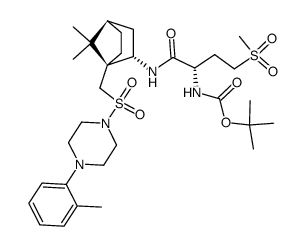
L-368,899 hydrochloride structure
|
Common Name | L-368,899 hydrochloride | ||
|---|---|---|---|---|
| CAS Number | 148927-60-0 | Molecular Weight | 591.22600 | |
| Density | 1.31g/cm3 | Boiling Point | N/A | |
| Molecular Formula | C26H43ClN4O5S2 | Melting Point | N/A | |
| MSDS | N/A | Flash Point | N/A | |
Use of L-368,899 hydrochlorideL-368,899 is an orally active and selective OT (oxytocin ) receptor antagonist, with IC50s of 8.9 and 26 nM for uterus of rat and human, respectively. L-368,899 can cross the blood-brain barrier (BBB). L-368,899 inhibits oxytocin-stimulated uterine contractions in rats and can be used in study of preterm labor[1][2][3]. |
| Name | L-368,899 hydrochloride,(2S)-2-Amino-N-[(1S,2S,4R)-7,7-dimethyl-1-[[[4-(2-methylphenyl)-1-piperazinyl]sulfonyl]methyl]bicyclo[2.2.1]hept-2-yl]-4-(methylsulfonyl)butanamide |
|---|---|
| Synonym | More Synonyms |
| Description | L-368,899 is an orally active and selective OT (oxytocin ) receptor antagonist, with IC50s of 8.9 and 26 nM for uterus of rat and human, respectively. L-368,899 can cross the blood-brain barrier (BBB). L-368,899 inhibits oxytocin-stimulated uterine contractions in rats and can be used in study of preterm labor[1][2][3]. |
|---|---|
| Related Catalog | |
| Target |
IC50: 8.9 nM (rat uterus), 26 nM (human uterus)[3]. |
| In Vivo | L-368,899 (0.1, 0.3, 1 mg/kg; infused i.v.; single) shows a dose-related antagonism of OT-stimulated uterine contractions with an AD50 value of 0.35 mg/kg in vivo[1]. L-368,899 (3, 10, 30 mg/kg; i.d.; single) inhibits the contractile effects of OT (AD50= 7 mg/kg) with a long (>4 h) duration of action in vivo (AD50: the dose of L-368,899 required to reduce the response to OT by 50%)[1]. L-368,899 (10 mg/kg, p.o.; single) shows bioavailability (AUC 0-6 h) of 35%[1]. L-368,899 (0.54, 1.8, 5.4 mg/kg; i.v.; single) reduces both oxytocin-induced and endogenous increases in plasma PGFM concentration[2]. Animal Model: Adult female Sprague-Dawley rats (250-350 g)[1]. Dosage: 0.1, 0.3, 1 mg/kg Administration: Infused intravenous injection; single. Result: Inhibited OT-stimulated uterine contractions with an AD50 value of 0.35 mg/kg. Animal Model: Adult female Sprague-Dawley rats (250-350 g)[1]. Dosage: 3, 10, 30 mg/kg Administration: Intraduodenal; single. Result: Exhibited a antagonism of OT-stimulated uterine contractions with an AD50 of 7 mg/kg and duration of action more than 4 h. Animal Model: Adult female Sprague-Dawley rats (250-350 g)[1]. Dosage: 10 mg/kg Administration: Oral administration, single. Result: Showed orally active with bioavailability (AUC 0-6 h) of 35%. Animal Model: Mature Dorset cross ewes (53-57 kg; Removal of ovaries)[2]. Dosage: 0.54, 1.8, 5.4 mg/kg (3, 10 and 30 µg/kg/min for 3 h; dissolved in 0.9% saline). Administration: Intravenous infusion; single. Result: Led to a significant decrease in both the frequency (from 2.2 to 1.0 episodes/ewe) and amplitude (from 68.8 to 31.8 pg/mL) of episodes of increased plasma concentration of PGFM. |
| References |
| Density | 1.31g/cm3 |
|---|---|
| Molecular Formula | C26H43ClN4O5S2 |
| Molecular Weight | 591.22600 |
| Exact Mass | 590.23600 |
| PSA | 146.64000 |
| LogP | 5.57760 |
| Index of Refraction | 1.61 |
| InChIKey | MWIASLNTAGRGGA-ZJPWWDJASA-N |
| SMILES | Cc1ccccc1N1CCN(S(=O)(=O)CC23CCC(CC2NC(=O)C(N)CCS(C)(=O)=O)C3(C)C)CC1 |
| Precursor 1 | |
|---|---|
| DownStream 0 | |
| selective oxytocin receptor antagonist |