
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:NMDA receptor antagonist 2
- 纯度:98.0%
- 规格:
- 联系人:徐经理

 351666998
复制
351666998
复制


 133 1186 9306
复制
133 1186 9306
复制

- 联系电话:133 1186 9306
查看所有供应商和价格请点击:
875898-41-2
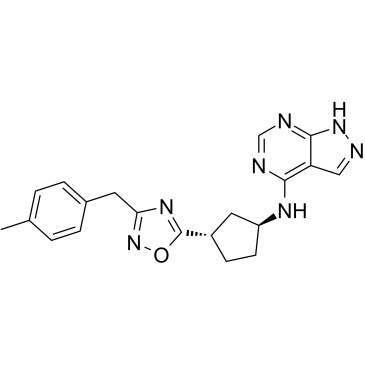
875898-41-2结构式
- 常用中文名:NMDA receptor antagonist 2
- 常用英文名:NMDA receptor antagonist 2
- CAS号:875898-41-2
- 分子式:C20H21N7O
- 分子量:375.43
- 相关类别: 信号通路 跨膜转运 iGluR
- 发布时间:2020-08-09 17:32:25
- 更新时间:2025-08-21 15:24:59
-
NMDA receptor antagonist 2 是一种有效的口服 NR2B 亚型选择性 NMDA 拮抗剂,其 IC50 和 Ki 分别为 1.0 nM 和 0.88 nM。NMDA receptor antagonist 2 用于神经性疼痛与帕金森病的研究。
| 英文名 | NMDA receptor antagonist 2 |
|---|
| 描述 | NMDA receptor antagonist 2 是一种有效的口服 NR2B 亚型选择性 NMDA 拮抗剂,其 IC50 和 Ki 分别为 1.0 nM 和 0.88 nM。NMDA receptor antagonist 2 用于神经性疼痛与帕金森病的研究。 |
|---|---|
| 相关类别 | |
| 靶点 |
IC50: 1.0 nM; Ki: 0.88 nM (NR2B subtype NMDA)[1] |
| 体外研究 | NMDA受体拮抗剂2在NR2B(Ki=0.88nm)具有良好的效价,对hERG结合具有选择性(IP=20000 nM)。NMDA受体拮抗剂2在表达重组NR1/NR2B受体的细胞中测定Ca2+流量。在MK-499-结合分析中评估hERG通道的选择性[1]。NMDA受体拮抗剂2在使用表达NR2B的细胞(IC50=1.0nm)的功能性试验中具有很强的效力,并且在使用均质化的人类颞叶皮质样品(Ki=0.81nm)的结合分析中保持等电位。在使用NR2B受体的电生理分析中,化合物22显示出完全阻断离子通量,KD=0.35nm。化合物22对NR2A(IC50=200μM)、hERG结合(IP=20μM)、基于哌唑嗪结合的α-肾上腺素能受体(IC50>100μM)和CYP P450(包括CYP3A4、2C9和2D6)也具有高水平的选择性[1]。 |
| 体内研究 | 在高等物种的药代动力学研究中,NMDA受体拮抗剂2在犬体内表现出良好的口服生物利用度(F=83%)、半衰期(T1/2=7.5小时)和清除率(CL=3.6毫升/分钟/千克)。在恒河猴体内具有中等清除率(CL=12ml/min/kg)和口服生物利用度(F=17%),半衰期(T1/2)为1.5小时[1]。在大鼠药代动力学研究中,NMDA受体拮抗剂2表现出口服生物利用度(F=23%)、半衰期(T1/2=0.7小时)和清除率(CL=24 mL/min/kg),大鼠口服NMDA受体拮抗剂ED50为4.8 mg/kg[1]。在神经源性疼痛大鼠脊髓神经结扎模型中,手术结扎脊柱内的两条腰神经可诱发机械性痛觉超敏状态[1]。NMDA受体拮抗剂2(口服;3-30mg/kg)在口服10和30mg/kg剂量后以剂量依赖的方式抑制触觉超敏。与药物治疗的动物相比,其最大可能效果平均提高15%(3 mg/kg)、41%(10 mg/kg)和69%(30 mg/kg)[1]。NMDA受体拮抗剂2(口服;3-30mg/kg)对帕金森病的急性啮齿动物模型有效。氟哌啶醇(HY-14538)的给药剂量已被证明能在大鼠中引起急性兴奋性反应,化合物22以剂量依赖性的方式降低了催化反应评分,与溶媒组相比,化合物22的平均改善率分别为34%(3 mg/kg)、86%(10 mg/kg)和92%(30 mg/kg)[1]。 |
| 参考文献 |
| 分子式 | C20H21N7O |
|---|---|
| 分子量 | 375.43 |