| Description |
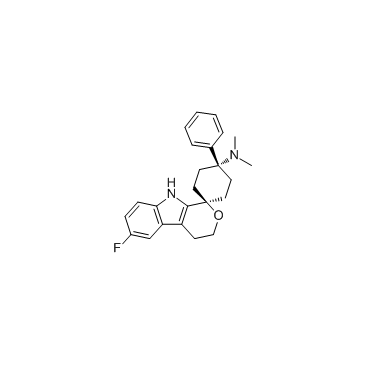
Cebranopadol is an analgesic NOP and opioid receptor agonist with Kis/EC50s of 0.9 nM/13 nM, 0.7 nM/1.2 nM, 2.6 nM/17 nM, 18 nM/110 nM for human NOP, MOP, KOP and delta-opioid peptide (DOP) receptor, respectively.
|
| Related Catalog |
|
| Target |
EC50: 13±2 nM (hNOP receptor), 1.2±0.4 nM (hMOP receptor), 17±5 nM (hKOP receptor), 110±28 nM (hDOP receptor)[1]
|
| In Vitro |
Cebranopadol binds with high affinity (subnanomolar to nanomolar range) to nociceptin/orphanin FQ peptide (NOP) and opioid receptors, with Ki of 1±0.5 nM, 2.4±1.2 nM, and 64±11 nM for rat NOP, mu-opioid peptide (MOP) receptor, and kappa-opioid peptide (KOP) receptor, and with Ki of 0.9±0.2 nM, 0.7±0.3 nM, and 2.6±1.4 nM for Rat NOP, MOP, and KOP receptor[1].
|
| In Vivo |
Cebranopadol exhibits highly potent and efficacious antinociceptive and antihypersensitive effects in several rat models of acute and chronic pain (tail-flick, rheumatoid arthritis, bone cancer, spinal nerve ligation, diabetic neuropathy) with ED50 values of 0.5-5.6 μg/kg after intravenous and 25.1 μg/kg after oral administration. In comparison with selective MOP receptor agonists, cebranopadol is more potent in models of chronic neuropathic than acute nociceptive pain. Cebranopadol’s duration of action is long (up to 7 hours after intravenous 12 μg/kg; >9 hours after oral 55 μg/kg in the rat tail-flick test). The antihypersensitive activity of cebranopadol in the spinal nerve ligation model is partially reversed by pretreatment with the selective NOP receptor antagonist J-113397 or the opioid receptor antagonist naloxone, indicating that both NOP and opioid receptor agonism are involved in this activity. Development of analgesic tolerance in the chronic constriction injury model is clearly delayed compared with that from an equianalgesic dose of morphine (complete tolerance on day 26 versus day 11, respectively). Unlike morphine, cebranopadol did not disrupt motor coordination and respiration at doses within and exceeding the analgesic dose range. Cebranopadol, by its combination of agonism at NOP and opioid receptors, affords highly potent and efficacious analgesia in various pain models with a favorable side effect profile[1].
|
| Kinase Assay |
Human MOP, DOP, KOP, and NOP receptor binding assays are run in microtiter plates with wheat germ agglutinin-coated scintillation proximity assay beads. [N-allyl-2,3-3H]naloxone and [tyrosyl-3,5-3H]deltorphin II, [3H]Ci-977, and [leucyl-3H]nociceptin are used as ligands for the MOP, DOP, KOP, and NOP receptor binding studies, respectively. The KD values of the radioligands used for the calculation of Ki values are provided as supplemental information. The assay buffer used for the MOP, DOP, and KOP receptor binding studies is 50 mM Tris-HCl (pH 7.4) supplemented with 0.052 mg/mL bovine serum albumin. For the NOP receptor binding studies, the assay buffer used is 50 mM HEPES, 10 mM MgCl2, 1 mM EDTA (pH 7.4). The final assay volume of 250 μL/well included 1 nM [3H]naloxone, 1 nM [3H]deltorphin II, 1 nM [3H]Ci-977, or 0.5 nM [3H]nociceptin as a ligand and cebranopadol in dilution series. Cebranopadol is diluted with 25% DMSO in water to yield a final 0.5% DMSO concentration, which also served as a respective vehicle control. Assays are started by the addition of beads (1 mg beads/well), which had been preloaded for 15 minutes at room temperature with 23.4 μg of human MOP membranes, 12.5 μg of human DOP membrane, 45 μg of human KOP membranes, or 25.4 µg of human NOP membranes per 250 µL of final assay volume. After short mixing, the assays are run for 90 minutes at room temperature. The microtiter plates are then centrifuged for 20 minutes at 500 rpm, and the signal rate is measured by means of a 1450 MicroBeta Trilux. IC50 values reflecting 50% displacement of [3H]naloxone-, [3H]deltorphin II-, [3H]Ci-977-, or [3H]nociceptin-specific receptor binding are calculated by nonlinear regression analysis. Individual experiments are run in duplicate and are repeated three times in independent experiments[1].
|
| Animal Admin |
Rats[1] The pharmacokinetic properties of cebranopadol in rats are investigated after a single intravenous dose of 160 μg/kg cebranopadol. The intravenous dose is administered as a bolus in a volume of 2 mL/kg with a catheter in the vena femoralis. Blood samples (200 μL/sample) are withdrawn via an implanted arterial catheter (arteria carotis) by an automated blood sampling system at the following sampling times: 0 (predose), 5, 15, 30, 60, 180, 360, 720, and 1440 minutes after administration. Blood samples are centrifuged, and plasma is separated. Plasma concentrations of cebranopadol are determined using a validated liquid chromatography-tandem mass spectrometry method. The lower limit of quantification for cebranopadol in this method is 0.05 ng/mL using a sample volume of 50 µL of plasma.
|
| References |
[1]. Linz K, et al. Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther. 2014 Jun;349(3):535-48. [2]. de Guglielmo G, et al. Cebranopadol Blocks the Escalation of Cocaine Intake and Conditioned Reinstatement of Cocaine Seeking in Rats. J Pharmacol Exp Ther. 2017 Sep;362(3):378-384. [3]. Satat K, et al. Evaluation of cebranopadol, a dually acting nociceptin/orphanin FQ and opioid receptor agonist in mouse models of acute, tonic, and chemotherapy-induced neuropathic pain. Inflammopharmacology. 2017 Oct 25.
|