BI 224436
更新时间:2025-08-25 06:55:02
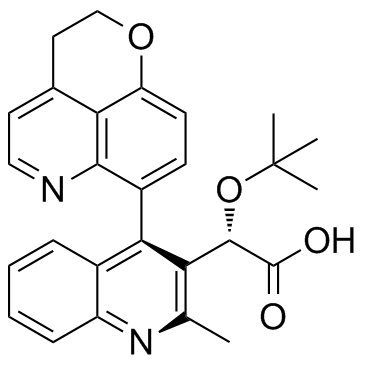
BI 224436结构式

|
常用名 | BI 224436 | 英文名 | BI 224436 |
|---|---|---|---|---|
| CAS号 | 1155419-89-8 | 分子量 | 442.506 | |
| 密度 | 1.3±0.1 g/cm3 | 沸点 | 631.8±55.0 °C at 760 mmHg | |
| 分子式 | C27H26N2O4 | 熔点 | N/A | |
| MSDS | N/A | 闪点 | 335.9±31.5 °C |
BI 224436用途BI 224436是一种新型的HIV-1非催化性位点整合酶抑制剂,对HIV-1实验室菌株的EC50值小于15 nM。 |

| 中文名 | BI 224436 |
|---|---|
| 英文名 | BI 224436 |
| 英文别名 | 更多 |
| 描述 | BI 224436是一种新型的HIV-1非催化性位点整合酶抑制剂,对HIV-1实验室菌株的EC50值小于15 nM。 |
|---|---|
| 相关类别 | |
| 靶点实验 |
EC50: 15 nM (HIV-1)[1] |
| 体外研究 | BI 224436具有超过90μM的细胞毒性。在50%人血清存在下,BI 224436的抗病毒效力降低了2.1倍。 BI 224436保留针对编码INSTI抗性取代N155S,Q148H和E92Q的重组病毒的完全抗病毒活性。 BI 224436与大多数批准的抗逆转录病毒药物(包括INSTI)一起显示出累加效应。 BI 224436具有类似药物的体外吸收,分布,代谢和排泄(ADME)特性,包括Caco-2细胞通透性,溶解度和低细胞色素P450抑制[1]。 |
| 体内研究 | BI 224436在大鼠中表现出优异的药代动力学特征(清除率为肝流量[CL],0.7%;生物利用度[F],54%),猴子(CL,23%; F,82%)和狗(CL, 8%; F,81%)[1]。 |
| 激酶实验 | 将BI 224436溶解在乙腈 - 甲醇(50:50,体积/体积)中以达到1.5mM的浓度。将磷酸盐缓冲液(pH 7.4),辅因子和测试物质或同种型选择性抑制剂加入到96孔板中,并预热至37℃10分钟。辅因子浓度为1.3mM NADP,3.3mM葡萄糖-6-磷酸和0.4U / mL葡萄糖-6-磷酸脱氢酶。通过加入预热(37℃)酶和底物引发反应。将反应混合物在37℃温育,并通过加入0.038ml 40:40:20(体积/体积)甲醇 - 乙腈-0.5M Tris缓冲液终止反应混合物。使用微孔板分光荧光计在特定的激发和发射波长下测量荧光代谢物的形成。 IC50使用SAS软件[1]提供的96孔32程序确定。 |
| 动物实验 | 大鼠:对于口服PK研究,BI 224436以0.5%(wt / vol)甲基纤维素(MC),0.3%(vol / vol)Tween 80和1%(vol / vol)N-甲基 - 的悬浮液给药。 2-吡咯烷(MP)在水中。对于iv给药,将BI 224436溶于70%PEG 400-30%水(体积/体积)中。将适量的BI 224436用超声处理溶解在PEG 400中。大鼠通过颈静脉接受单次iv剂量的0.2mg / kg体重(1mL / kg)作为推注或接受通过管饲法施用的单次口服剂量0.4mg / kg(10mL / kg)。在给药后0,0.25,0.5,1,1.5,2,3,4,6,8,12,24和32小时获得血样用于分析[1]。 |
| 参考文献 |
| 密度 | 1.3±0.1 g/cm3 |
|---|---|
| 沸点 | 631.8±55.0 °C at 760 mmHg |
| 分子式 | C27H26N2O4 |
| 分子量 | 442.506 |
| 闪点 | 335.9±31.5 °C |
| 精确质量 | 442.189270 |
| LogP | 4.11 |
| 蒸汽压 | 0.0±1.9 mmHg at 25°C |
| 折射率 | 1.661 |
| 储存条件 | 2-8℃ |
共94条,当前第1页,共10页
1
2
3
4
5
| 99A996378Y |
| BI 224436 |
| (2S)-[4-(2,3-Dihydropyrano[4,3,2-de]quinolin-7-yl)-2-methyl-3-quinolinyl][(2-methyl-2-propanyl)oxy]acetic acid |
本网页内容来自不同专业数据源,如对内容有疑义,欢迎联系service1@chemsrc.com。

我想出现在这里:
 351666998
复制
351666998
复制

 13311869306
复制
13311869306
复制

- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:BI 224436
- 纯度:99.0%
- 价格: ¥15000.9/50mg ¥5000.9/10mg ¥3500.9/5mg ¥2333.9/2mg
- 联系人:阿拉丁
 2881459338
复制
2881459338
复制
- 联系电话:400-620-6333
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:BI-224436
- 纯度:98.0%
- 价格:
- 联系人:徐经理
351666998
复制
 13311869306
复制
13311869306
复制
- 联系电话:13311869306
查看所有供应商和价格请点击: