869490-23-3
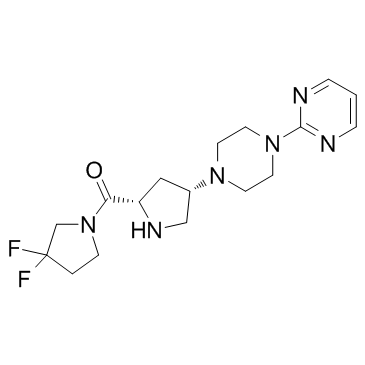
869490-23-3 structure
- Name: Gosogliptin
- Chemical Name: (3,3-difluoropyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-ylpiperazin-1-yl)pyrrolidin-2-yl]methanone
- CAS Number: 869490-23-3
- Molecular Formula: C17H24F2N6O
- Molecular Weight: 366.40900
- Catalog: Signaling Pathways Metabolic Enzyme/Protease Dipeptidyl Peptidase
- Create Date: 2016-02-15 13:43:46
- Modify Date: 2026-07-14 08:52:32
-
Gosogliptin is a potent and selective inhibitor of dipeptidyl peptidase-IV (DPP-IV).
| Name | (3,3-difluoropyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-ylpiperazin-1-yl)pyrrolidin-2-yl]methanone |
|---|---|
| Synonyms |
unii-gi718uo477
gosogliptin |
| Description | Gosogliptin is a potent and selective inhibitor of dipeptidyl peptidase-IV (DPP-IV). |
|---|---|
| Related Catalog | |
| Target |
DPP-IV[1][2] |
| In Vitro | Gosogliptin (PF-00734200) is a potent, orally active, selective, and competitive inhibitor of DPP-IV, the enzyme mainly responsible for the degradation of the incretin peptides GLP-1 and glucose-dependent insulinotropic polypeptide. Gosogliptin demonstrates greater than 200-fold selectivity over other members of the DPP family (DPP-2, DPP-3, DPP-8, and DPP-9) and the related serine proteases, fibroblast activation protein, aminopeptidase P, and propyl oligopeptidase, enzymes that possess similar catalytic activities. Gosogliptin demonstrates rapid and reversible inhibition of plasma DPP-4 activity when administered orally to rats, dogs, and monkeys. In various nonclinical models, Gosogliptin stimulates insulin secretion and improves glucose tolerance[2]. |
| In Vivo | The objectives of the present study are to characterize the metabolism, pharmacokinetics, and excretion of [14C] Gosogliptin in Sprague-Dawley (SD) rats, beagle dogs, and humans. A single dose of [14C] Gosogliptin is administered orally to intact SD rats (5 mg/kg), beagle dogs (5 mg/kg), and humans (20 mg). After a single oral dose of [14C] Gosogliptin to SD rats, an overall mean of 97.1% of the administered radioactivity is recovered in the urine, feces, and cage wash over a period of 168 h postdose. The mean cumulative dose recovered in feces is 66.0%. The mean cumulative excretion in the urine is 30.8%. Approximately 95% of the excreted radioactivity recovery occurred in the first 48 h. Mean total recoveries of dosed radioactivity from bile duct-cannulated rats are 29.5% in urine and 62.0% in bile. No gender-related differences are observed in the excretion pattern of radioactivity[2]. |
| Kinase Assay | Studies to identify the P450 isoform(s) responsible for formation of M5 are conducted using chemical inhibition and incubation in recombinant P450 isozymes. Inhibition studies are performed with each of the following inhibitors: Furafylline (10 μM for CYP1A2), Sulfaphenazole (10 μM for CYP2C9), Quinidine (10 μM for CYP2D6), (+)N-3-benzylnirvanol (10 μM for CYP2C19), and Ketoconazole (1 μM for CYP3A4). Incubations (1 ml) are performed in duplicate with NADPH (1.3 mM) in 1.5 mL plastic Eppendorf tubes open to air at 37°C in a shaking water bath. Samples are preincubated at 37°C for 5 min before the addition of NADPH. Each incubation contains microsomes (2 mg/mL protein), 100 mM potassium phosphate buffer (pH 7.4), 10 mM MgCl2, 10 μM Gosogliptin (PF-00734200), and one of the above inhibitors. A control sample is also prepared without inhibitor. Additional controls using marker substrates (each at 10 μM), in the presence and absence of P450-specific inhibitors, are used to confirm inhibition results and included Phenacetin (CYP1A2), Tolbutamide (CYP2C9), (S)-Mephenytoin (CYP2C19), Dextromethorphan (CYP2D6), and Testosterone (CYP3A4). At 0 and 30 min, incubations are quenched with an equal volume of ice-cold acetonitrile. Samples are then placed on ice for 15 min to allow precipitation of protein and subsequently centrifuged at 1800g for 5 min. An aliquot is removed from each sample and injected onto the HPLC/MS system[2]. |
| Animal Admin | Rats[2] A group of SD rats (n=3/gender) is administered a single 5 mg/kg oral dose of [14C] Gosogliptin for the mass balance study. For biliary excretion experiments, another group of two male and two female bile duct-cannulated rats is administered a single 5 mg/kg oral dose of [14C] Gosogliptin in similar fashion. The dose is formulated as a suspension in 0.5% methyl cellulose on the day before dose administration. Each rat received an approximate dose of ∼60 μCi of radiolabeled material. Urine and feces are collected from intact animals for 7 days at 0 to 8, 8 to 24, 24 to 48, 48 to 72, 72 to 96, 96 to 120, 120 to 144, and 144- to 168-h intervals after the dose. The first feces sample is collected at 0 to 24 h after the dose. Bile and urine samples are collected from bile duct-cannulated animals at 0- to 8-, 8- to 24-, and 24- to 48-h intervals after the dose. The volumes of urine and bile samples are recorded, and all of the biological samples are stored at -20°C until analysis. For determination of pharmacokinetic parameters and identification of circulating metabolites, a third group of jugular vein-cannulated rats (n=10/gender) is given an oral dose of 5 mg/kg [14C] Gosogliptin. Blood from two animals per gender is collected at 0.5, 1, 2, 4, and 8 h postdose in heparinized (lithium heparin anticoagulant) tubes. The blood samples are centrifuged at 1000g for 10 min to obtain the plasma. Plasma is transferred to clean tubes and stored at -20°C until analysis. |
| References |
| Molecular Formula | C17H24F2N6O |
|---|---|
| Molecular Weight | 366.40900 |
| Exact Mass | 366.19800 |
| PSA | 64.60000 |
| LogP | 0.46630 |