
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:Cariprazine
- 纯度:98.0%
- 规格:25mg/50mg/5mg/1g
- 联系人:阿拉丁

 2881459338
复制
2881459338
复制
- 联系电话:400-620-6333
- 上海阿摩尔生物科技有限公司
- 上海市 上海市 奉贤区
- 产品名:3-(trans-4-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)环己基)-1,1-二甲基脲
- 纯度:99.0%
- 规格:100mg/10mg/1g/250mg
- 联系人:王汐瑶
3623107365
复制
- 联系电话:18916960931
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:Cariprazine
- 纯度:98.0%
- 规格:
- 联系人:徐经理
351666998
复制


 133 1186 9306
复制
133 1186 9306
复制

- 联系电话:133 1186 9306
查看所有供应商和价格请点击:
839712-12-8
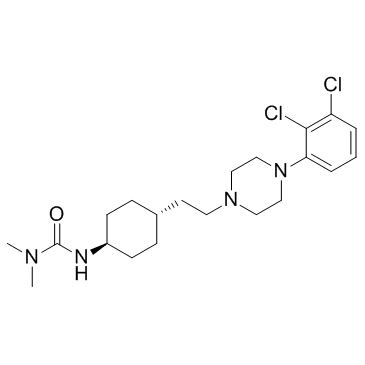
839712-12-8结构式
- 常用中文名:卡利拉嗪
- 常用英文名:Cariprazine
- CAS号:839712-12-8
- 分子式:C21H32Cl2N4O
- 分子量:427.411
- 相关类别: 信号通路 G 蛋白偶联受体/G 蛋白 5-HT受体
- 发布时间:2016-06-23 04:05:32
- 更新时间:2026-07-03 10:55:40
-
Cariprazine是一种新型抗精神病候选药物,结合D3, D2 和 5-HT1A 的 Ki 为0.085 nM,0.49 nM,2.6 nM。
| 中文名 | 卡利拉嗪 |
|---|---|
| 英文名 | 3-[4-[2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl]cyclohexyl]-1,1-dimethylurea |
| 英文别名 |
trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-1-yl]-ethyl}-N,N-dimethylcarbamoyl-cyclohexylamine
trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}-N,N-dimethylcarbamoyl-cyclohexylamine Cariprazine UNII-F6RJL8B278 Vraylar RGH 188 N'-(trans-4-{2-[4-(2,3-dichlorophenyl)piperazin-1-yl]ethyl}cyclohexyl)-N,N-dimethylurea 3-(trans-4-(2-(4-(2,3-dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-1,1-dimethylurea Cariprazine (USAN/INN) 3-(trans-4-{2-[4-(2,3-Dichlorophenyl)-1-piperazinyl]ethyl}cyclohexyl)-1,1-dimethylurea trans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea UNII:F6RJL8B278 |
| 描述 | Cariprazine是一种新型抗精神病候选药物,结合D3, D2 和 5-HT1A 的 Ki 为0.085 nM,0.49 nM,2.6 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
Ki: 0.49 nM (D2 receptor), 0.085 nM (D3 receptor), 2.6 nM (5-HT1A receptor)[1] |
| 体外研究 | Cariprazine以高效力(pEC50 8.5)刺激肌醇磷酸(IP)形成,效力相对较低(Emax 30%)[2]。 Cariprazine,一种新型候选抗精神病药物,对人类D3与人D2L和人D2S受体的亲和力分别高出约10倍(分别为pKi 10.07,9.16和9.31)。卡立嗪对人血清素(5-HT)2B型受体(pKi 9.24)具有高亲和力,具有纯拮抗作用。卡利嗪对人和大鼠海马5-HT1A受体具有较低的亲和力(分别为pKi 8.59和8.34),并且表现出低的内在功效。 Cariprazine对人5-HT2A受体(pKi 7.73)表现出低亲和力。对组胺H1和5-HT2C受体的中度或低亲和力(分别为pKi 7.63和6.87)表明,与这些受体相关的不良反应倾向降低了卡立嗪[2]。在抑制异丙肾上腺素诱导的HEK-293细胞cAMP产生中,卡立嗪比阿立哌唑(EC50 = 9.2 nM)强6倍(EC50 = 1.4 nM)[4]。 |
| 体内研究 | 与基线PET测量相比,施用哌嗪(30μg/ kg)将两种放射性配体的纹状体摄取降低至非特异性结合水平。卡立嗪对小脑的时间-活动曲线的影响可忽略不计。在剂量为5.0和30μg/ kg时,对于拮抗剂[11C] raclopride和激动剂放射性配体[11C] MNPA,卡立哌嗪引起剂量依赖性多巴胺D2/D3受体占据约45%和约80%。使用瞬时平衡和MRTM2方法计算的多巴胺D2/D3受体的受体占有率在最低剂量(1.0μg/ kg)下为5%至最高剂量(300μg/ kg)下的94%[1]。研究5种剂量的卡利拉嗪(0.005至0.15mg/kg)对野生型小鼠的EPM行为的影响。虽然较低剂量的卡利拉嗪(0.005至0.02毫克/千克)不会改变开放臂的时间,但两个较高剂量(0.08和0.15毫克/千克)会导致该措施的显着下降(ANOVA,(F(5) ,52)= 4.20; p = 0.0032))。此外,两个较高剂量的卡立哌嗪也导致手臂入口总数显着减少(F(5,52)= 7.21; p = 0.0001))但这种手臂入口总数的减少主要归因于通过显着减少闭臂数(F(5,52)= 11.75; p = 0.0001))。两种最高剂量的卡利拉嗪(0.08和0.15 mg/kg)对运动活动有显着影响,但剂量范围为0.005至0.02 mg/kg不影响EPM试验中的焦虑样行为或运动活动[3]。急性ip给药所有剂量的Cariprazine后,观察到哇巴因诱导的机能亢进显着(P <0.01)减少(平均值±SEM:0.06 mg/kg,64.2±3.88; 0.25 mg/kg,72.7±11.67; 0.5 mg /与单独的哇巴因注射液(114.6±14.33)相比,kg,40.6±5.32; 1 mg/kg,19.5±8.78)和锂(40.4±12.78)。最高的卡利拉嗪剂量产生显着的镇静作用(对于卡利普拉嗪1.0mg/kg aCSF对生理盐水aCSF的抑制率为72%; P <0.05)[4]。 |
| 激酶实验 | 这些测定在50mM Tris(pH 7.4),100mM NaCl,7mM MgCl 2,1mM EDTA和1mM DTT中进行。分析管(终体积250μL)含有50μM(纹状体和海马)或1μM(D2和D3细胞膜)GDP,待检测的配体和膜悬浮液(250μg组织/管用于纹状体和海马体,20μL)用于hD2和hD3膜的μg蛋白质/管)。将样品在30℃下预孵育10分钟。加入50μM[35S]GTPγS后,将膜在30℃下再温育60分钟。在10μMGTPγS存在下测定非特异性结合;仅在缓冲液存在下测定基础结合。通过使用收获器通过UniFilter GF / B快速过滤终止测定,并且用1mL冰冷的缓冲液将膜洗涤四次。在干燥(40℃,1小时)后,将40μLMicroscint加入到过滤器中,并通过TopCount NXT计数器[2]确定结合的放射性。 |
| 细胞实验 | 将细胞接种在24孔组织培养板上的500μL培养基中。加入50微升含有0.55μCi肌 - [3H]肌醇的培养基(终浓度1μCi/ mL)并孵育18-20小时。然后用含有140mM NaCl,5mM KCl,2mM CaCl 2,5mM HEPES,5mM Na-HEPES,20mM葡萄糖和10mM LiCl(pH 7.4)的缓冲液洗涤细胞三次。然后将细胞在仅含有测试化合物(激动剂测试)或与1000nM(±)-Quinpirole(拮抗剂测试)一起的培养基中再孵育60分钟(37℃)。然后吸出培养基,通过加入400μL0.1MHCl / 2mM CaCl2裂解细胞,并将上清液在-72℃冷冻。解冻并以1000g离心10分钟后,将200μL每种上清液上样到250μLAG1-X8(甲酸盐形式)阴离子交换柱上。弃去流出物,并将柱在1.5mL蒸馏水中洗涤两次。将IP用2.5mL 1M甲酸铵/0.1M甲酸直接洗脱到闪烁瓶中,加入10mL Optiphase HiSafe 3,并在TriCarb 4900闪烁计数器中测定放射性[2]。 |
| 动物实验 | 小鼠[3]在野生型C57Bl / 6J小鼠上进行实验。在认知功能的测试中,必须使用对情绪行为没有影响且不损害运动活动的药物浓度。首次测试的卡吡嗪(剂量范围为0.005至0.15mg / kg)是否影响EPM中小鼠的行为,这是一种焦虑相关行为的测试,其也严重依赖于正常的运动活动。将动物暴露于为小鼠设计的EPM装置(腿高:45cm,臂长:35cm,泳道宽度:5cm,壁高:15cm)。在下午1点到4点之间进行测试(低于100勒克斯照明)。将小鼠置于迷宫的中心,并将它们的时间花在张开的臂上,记录5分钟测试期间的闭合和开放臂入口的数量。张开双臂花费的时间和张开臂的数量的测量可以作为焦虑行为的衡量标准。闭合臂入口的数量用作运动活动的量度。大鼠[4]使用成年雄性Sprague-Dawley大鼠(150-300g)。将卡利拉嗪溶于0.9%盐水中,并在icv注射哇巴因前1小时通过腹膜内(ip)注射以0.06,0.25,0.5和1.0mg / kg施用,之后每天注射7天。在icv注射后立即评估开放野外活动,并在7天后再次评估(在最后一次ip注射卡利拉嗪后10-14小时注意到活性)。 |
| 参考文献 |
| 密度 | 1.2±0.1 g/cm3 |
|---|---|
| 沸点 | 600.1±55.0 °C at 760 mmHg |
| 分子式 | C21H32Cl2N4O |
| 分子量 | 427.411 |
| 闪点 | 316.7±31.5 °C |
| 精确质量 | 426.195313 |
| PSA | 42.31000 |
| LogP | 5.18 |
| 蒸汽压 | 0.0±1.7 mmHg at 25°C |
| 折射率 | 1.595 |
| 储存条件 | -20°C |
|
~95%
839712-12-8 |
| 文献:WO2011/73705 A1, ; Page/Page column 10 ; |
|
~%
839712-12-8 |
| 文献:Journal of Medicinal Chemistry, , vol. 56, # 22 p. 9199 - 9221 |
|
~%
839712-12-8 |
| 文献:Journal of Medicinal Chemistry, , vol. 56, # 22 p. 9199 - 9221 |
|
~%
839712-12-8 |
| 文献:Journal of Medicinal Chemistry, , vol. 56, # 22 p. 9199 - 9221 |
|
~%
839712-12-8 |
| 文献:Journal of Medicinal Chemistry, , vol. 56, # 22 p. 9199 - 9221 |
|
~%
839712-12-8 |
| 文献:Journal of Medicinal Chemistry, , vol. 56, # 22 p. 9199 - 9221 |

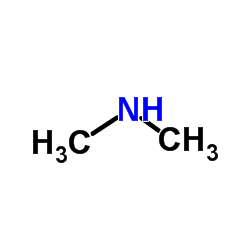
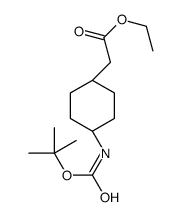
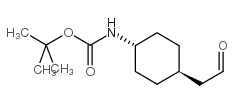
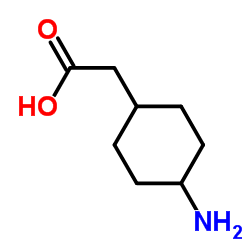
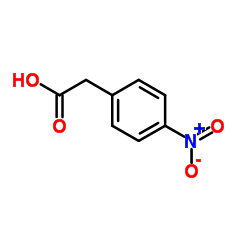
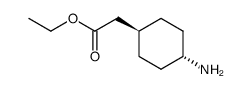
| 上游产品 6 | |
|---|---|
| 下游产品 1 | |
